 Рейтинг: 4.0/5.0 (1901 проголосовавших)
Рейтинг: 4.0/5.0 (1901 проголосовавших)Категория: Руководства

Процессы развития фармацевтической отрасли в России требуют гармонизации национального законодательства с требованиями Европейского Союза, разработки соответствующей нормативно-правовой базы, а также внедрения на предприятиях и организациях отрасли международных стандартов, прежде всего надлежащей производственной практики (GMP).
Данное Руководство должно послужить основой подготовки новых нормативно-правовых документов, которые будут касаться всех стадий обращения лекарственных средств.
Рекомендовано к использованию Федеральной службой по надзору в сфере здравоохранения и социального развития специалистам, занятым в сфере обращения лекарственных средств.
I. Руководство по валидации методик анализа лекарственных средств (методические рекомендации).
II. Руководство по составлению сведений о производстве лекарственных препаратов для включения в регистрационное досье (методические рекомендации).
III. Руководство по составлению сведений о валидации процесса производства лекарственных препаратов для включения в регистрационное досье (методические рекомендации).
М. Литтерра, 2008. — 132 c. Типовое руководство предприятия по производству лекарственных средств, подготовленное Федеральным союзом фармпроизводителей Германии (ВАН), 2004. Содержание: Предисловие редактора русского издания. Предисловие. Введение. Термины и определения. Интерпретация руководств СРМР/1СН 02А и 02В по валидации аналитических методик.
М. Изд-во «Перо», 2014. - 656 с. ил. ISBN 978-5-91940-743-0 Книга раскрывает многообразие и специфику инструментальных методов анализа как одного из ключевых методов оценки качества лекарственных препаратов. В издании приводится широкий перечень современных высокотехнологичных приборов с предельно четкими характеристиками, которые позволяют обеспечить получение правильных.
Быстрицкий Л.Д. Бикбаев А.А. Пикула Н.П. Дьяконова Е.В. Соляник Р.Г. Учебное пособие. — Томск: Изд-во ТПУ, 2011. — 258 с. В пособии рассмотрены вопросы государственного регулирования в сферах фармацевтических и биотехнологических процессов и производств, требования основных нормативных документов, регламентирующих проектирование, организацию и работу предприятий в.
12-е изд-е. — М. Научный центр экспертизы средств медицинского применения, 2008. — 704 с. — ISBN 978-5-9901447-1-2. OCR Общие Фармакопейные и Фармакопейные статьи, включенные в настоящее издание, утверждены приказом Минздравсоцразвития России от 15 октября 2007 г. № 641-Р85 Двенадцатому изданию Государственной Фармакопеи Российской Федерации предшествовали следующие издания.
Женева, 1999. - 165 с. В Руководстве дан обзор типов валидации и обозначен объём продиктованных GMP мер по обеспечению валидации; рассмотрены вопросы подготовки основного плана мероприятий по валидации; представлены форматы протоколов валидации технологического оборудования и производственных систем, а также форматы протоколов валидации технологического процесса и результатов.
M. АСИНКОМ, 2012. — 583 c. В монографии дается подробный анализ правил GMP. Федотов Александр Евгеньевич, доктор технических наук, президент Ассоциации инженеров по контролю микрозагрязнений (АСИНКОМ), главный редактор журнала "Технология чистоты", председатель технических комитетов по стандартизации ТК 184 "Обеспечение промышленной чистоты" и ТК 458 "Производство и контроль.


Дана оценка требований к валидации биоаналитических методик в соответствии с документами U.S. FDA и Европейского Медицинского Агентства (EMA).
В документах U.S. FDA и EMA биоаналитической методикой называется методика количественного определения лекарственных веществ (ЛВ) и (или) их метаболитов в биологических объектах (цельной крови, плазме, сыворотке, моче и др.) человека и животных [1,2]. Биоаналитические методики используются в основном при проведении фармакокинетических исследований (в т.ч. исследований биоэквивалентности), токсикокинетических исследований, изучении метаболизма. Основные методы анализа, применяемые для определения ЛВ и их метаболитов в биологических жидкостях, преимущественно относятся к хроматографическим (ВЭЖХ, ГЖХ, ВЭЖХ-МС, ВЭЖХ-МС-МС, ГХ-МС, ГХ-МС-МС), а также к иммунологическим (например, твердофазный иммуноферментный анализ или поляризационно-флуоресцентный иммуноанализ) и микробиологическим [1,2].
Согласно рекомендациям ICH [3,4] биоаналитическая методика, как и любая аналитическая методика, должна быть подвергнута валидации. При этом следует отметить, что требования к валидации фармакопейных аналитических и биоаналитических методик различаются [5]. Это, в первую очередь, связано с большей вариабельностью результатов биоаналитических определений (поскольку такие методики могут включать в себя процессы экстракции, осаждения белков и др.), а также со сложным составом биологической матрицы, содержащей определяемое вещество [6].
Особенности валидации биоаналитических методик
В настоящее время существуют два основных руководства по валидации биоаналитических методик – руководство для предприятий U.S. FDA 2001 г. [1] и драфт-руководство Европейского Медицинского Агентства (ЕМА) 2009 г. [2]. В Российской Федерации нормативные документы, регламентирующие процедуру подобной валидации, отсутствуют, а данные вопросы затрагиваются в научных публикациях. Валидация биоаналитической методики может быть полной, частичной либо кросс-валидацией. Полную валидацию проводят при разработке новой биоаналитической методики. Частичную валидацию проводят при смене лаборатории, оборудования, условий хранения проб и т.д. Для проведения частичной валидации обычно бывает достаточно провести определение правильности и прецизионности на уровнях intra-day (between-run) и, если применимо, определение стабильности. Кросс-валидацию проводят при необходимости сравнения результатов определений, полученных из разных лабораторий. При этом результаты анализа определяемого вещества с помощью обеих методик не должны различаться более чем на 15 % [1,2].
Валидационные характеристики биоаналитической методики. Специфичность/селективность (selectivity)
Специфичность – это способность методики безусловно определять действующее вещество в присутствии компонентов, которые могут содержаться в пробе [7]. Для определения специфичности методики проводят анализ не менее 6 образцов чистой биологической жидкости. Параллельно проводят анализ чистой биологической жидкости, к которой предварительно прибавляют стандартный раствор определяемого вещества или веществ (и внутреннего стандарта, если это применимо) в таком количестве, чтобы его содержание в пробе находилось в предполагаемом диапазоне концентраций фармакокинетической кривой.
Также анализируют стандартный раствор определяемого вещества и при необходимости – внутреннего стандарта. На всех хроматограммах образцов чистой биологической жидкости не должно наблюдаться пиков со временем удерживания, соответствующим времени удерживания определяемого вещества и внутреннего стандарта, которое определяют по хроматограммам стандартного раствора. Руководство ЕМА уточняет, что максимально допустимое мешающее воздействие посторонних веществ с временами удерживания определяемого вещества (и внутреннего стандарта, если это применимо) не должно превышать 20% от предела количественного определения (ПКО) определяемого вещества. В противном случае необходимо изменение условий методики определения [1,2].
Источник: журнал "Ремедиум" №12 (2011)
Валидационные характеристики и показатели точности. Объем валидации для фармакопейных и нефармакопейных методик испытаний. Особенности валидации микробиологических методов контроля качества (микробиологической чистоты и стерильности). Документирование.
Слово «VALID » впервые было использовано в английском языке в середине 17 века и в переводе означало «валидный», т.е.:
- действительный, имеющий силу;
- веский, обоснованный;
- лежащий на надежной, логичной основе.
Т.е. валидировать – делать валидным, утверждать, обосновывать, придавать законную силу.
В середине 19 века в отчете Лондонского аптечного общества был описан прообраз валидации поставки материалов в виде деятельности «комитета по рассмотрению различных вопросов», который собирался каждую пятницу, чтобы подтвердить качество сырья, поставленного на предыдущей неделе. Сегодня оно используется для обозначения: Validation – 1) утверждение, ратификация; 2) легализация, придание законной силы 3) проверка работоспособности; 4) подтверждение, проверка истинности; и т.д.
Валидация – документально оформленные действия, которые в соответствии с принципами надлежащей производственной практики доказывают, что определенная процедура, процесс, деятельность или система приводят к ожидаемым результатам с заранее установленными критериями приемлемости (ТКП 030-2013. «Надлежащая производственная практика»)
Валидируются как процессы, так и методики испытаний.
Испытание – определение одной или нескольких характеристик объекта согласно процедуре
Методика испытаний (МИ)– документированная процедура, включающая совокупность операций и требований, выполнение которых обеспечивает определение количественных или качественных показателей объекта испытаний. Валидация методики испытания – документированное подтверждение обоснованности (правильности) выбора методики испытаний, гарантирующее получение ожидаемых и воспроизводимых результатов, соответствующих поставленной цели (ГФ РФ)
Цель валидации МИ – экспериментальное доказательство того, что методика пригодна для решения поставленных задач .
Виды валидации МИ:
• Перспективная валидация – осуществляется при разработке новой продукции до начала серийного производства ЛС;
•Сопутствующая валидация – осуществляется во время серийного производства продукции, если перспективная валидация при определенных обстоятельствах не была завершена или были незначительные изменения в процессе производства (изменение формы таблеток).
Порядок проведения валидации:
• Наличие необходимого и достаточного аттестованного лабораторного оборудования; • Наличие обученного персонала требуемой квалификации;
• При проведении валидации используются только стандартные образцы с известными характеристиками, подтвержденными документально;
• Соблюдение трех этапов валидации:
- разработка и утверждение плана валидации МИ
- проведение валидации в строгом соответствии с планом валидации. Каждая стадия валидации должна быть документально оформлена и утверждена
- подготовка отчета о валидации, с указанием всех замеченных отклонений и выводов с рекомендациями по устранению отклонений.
Типовой перечень валидационных характеристик и показателей точности, которые должны учитываться при проведении валидации МИ:
1. Показатели точности:
•Правильность
•Прецизионность (точность) -повторяемость (сходимость) -внутрилабораторная воспроизводимость -межлабораторная воспроизводимость
2. Валидационные характеристики:
•Специфичность
•Робастность
•Предел обнаружения
•Предел количественного определения
•Линейность
•Диапазон применения
Объем валидации МИ
• Валидация в полном объеме – если используемые МИ не включены в действующие ТНПА
• Валидация в сокращенном объеме – если используемые МИ включены в действующие ТНПА. Объем валидации в данном случае определяется поставленной целью. Все методики и испытания, включенные в фармакопеи стран-участников ICH (ICH - The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use – Международная конференция по гармонизации технических требований к регистрации лекарственных препаратов для использования у человека), являются валидированными.
Валидация фармакопейных методик может быть выполнена в меньшем объёме, так как в полном объёме это было выполнено ранее при разработке.
Цель такой валидации – подтверждение на основании экспериментальных данных, что данная лаборатория в состоянии корректно воспроизвести фармакопейную методику или испытание:
• На конкретном аналитическом оборудовании;
• При использовании данных реактивов;
• В данных условиях окружающей среды;
• При выполнении анализа аналитиками данной лаборатории.
Методики испытаний микробиологических показателей:
1. Количественное определение (активность)
2. Микробиологическая чистота
3. Стерильность
Каждое отдельное исследование оформляется в виде протокола.
Основные требования к протоколам:
• должны содержать информацию об условиях, в которых проводились испытания;
• должны в полном объеме содержать первичные данные
испытаний;
• должны последовательно отображать ход эксперимента.
Валидация методики определения количественного содержания (активности) антибиотиков микробиологическим методом диффузии в агар: прецизионность (повторяемость, внутрилабораторная воспроизводимость), правильность.
Валидация методики определения количественного содержания (активности) антибиотиков микробиологическим методом (диффузии в агар)
Количественное определение – предназначено для определения количества анализируемого вещества в образце.
Методика количественного определения антибиотиков микробиологическим методом – методика количественного определения действующего вещества.
Для валидации фармакопейной методики определения количественного содержания (активности) действующих веществ в антибиотиках, в т.ч. микробиологическим методом, обязательными параметрами являются показатели точности:
- прецизионность (точность); - правильность,
Прецизионность (точность).
• Прецизионность МИ - выражение степени близости результатов для серии измерений, выполненных по данной методике на различных пробах одного итого же образца (одной серии).
• Прецизионность подтверждается повторяемостью, внутри- и межлабораторной воспроизводимостью.
Повторяемость – характеризует точность методики при ее выполнении в течение небольшого промежутка времени в одних и тех же условиях: одним аналитиком, при неизменных условиях окружающей среды (t, W, P).
• Используют трехдозный метод диффузии в агар. Тест- микроорганизмы, питательные среды, буферные растворы – в соответствии с.
• Готовят чашки Петри, как для определения активности антибиотиков методом диффузии в агар трехдозным методом с использованием требуемого тест-микроорганизма
• Повторяемость оценивают по результатам 6-ти испытаний по 2 параллельных испытания для каждого из 3-х контейнеров (флаконов) одной серии препарата. 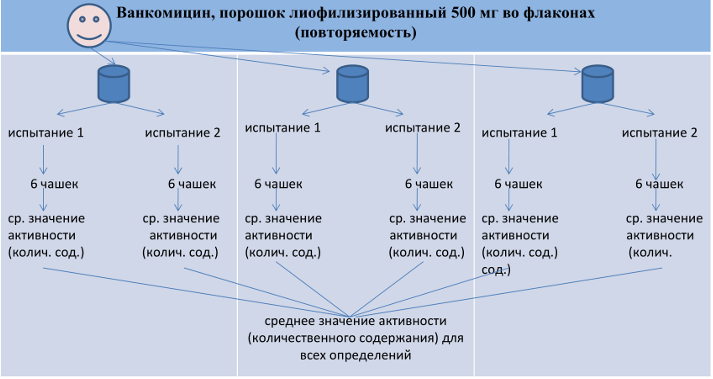
Находят:
- Стандартное отклонение для каждого определения;
- Среднее значение стандартного отклонения (для всех определений)
- Относительное стандартное отклонение (RSD)
• Критерий приемлемости для подтверждения повторяемости – величина относительного стандартного отклонения (RSD): отношение среднего значения стандартного отклонения к среднему значению активности (количественного содержания) исследуемого вещества для всех определений (%). • RSD – не более 2%.
Внутрилабораторная воспроизводимость - устанавливает влияние случайных факторов на точность валидируемой методики.
• Типичные исследуемые факторы: различные аналитики, различное оборудование, различные дни.
• Например: испытание проводится двумя аналитиками в разные дни. Каждый специалист проводит 6 испытаний для каждого из 3-х контейнеров (флаконов) одной серии препарата.
• Ход испытания – аналогично исследованию повторяемости. 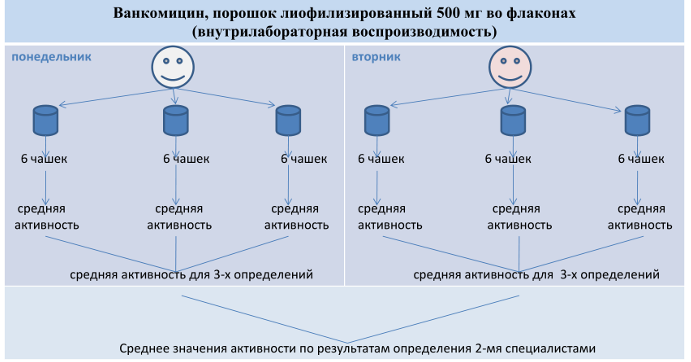
Находят:
- Стандартное отклонение для определения каждым специалистом;
- Среднее значение стандартного отклонения для определений двумя специалистами
- Относительное стандартное отклонение (RSD)
Критерий приемлемости для подтверждения внутрилабораторной воспроизводимости - величина относительного стандартного отклонения (RSD): отношение среднего значения стандартного отклонения к среднему значению активности (количественного содержания) по результатам определения двумя специалистами (%).
• RSD – не более 3%. Межлабораторная воспроизводимость – оценивается путем проведения межлабораторных исследований. (Необязательна для генерических препаратов). Правильность Правильность МИ - выражение степени соответствия между принятым эталонным значением и значением, полученным на основании большой серии результатов испытаний по данной методике. Принятое эталонное значение – например, теоретически рассчитанное значение содержания вещества в модельном растворе. Испытание проводят трехдозным методом как описано выше. Правильность оценивают не менее чем для 9 определений для трех различных концентраций вещества (80%, 100%, 130% от номинального содержания в ЛС или АФИ): по три определения (по три чашки) для каждой концентрации. Например: • ЛС ванкомицин, порошок лиофилизированный 500 мг во флаконах содержит 500 мг активного вещества (ванкомицина гидрохлорида - АФИ). 80% - 400 мг – образец №1 100% - 500 мг – образец №2 130% - 650 мг – образец №3. Готовят соответствующие модельные растворы АФИ трех концентраций и проводят определение трехдозным
методом диффузии в агар
Анализ результатов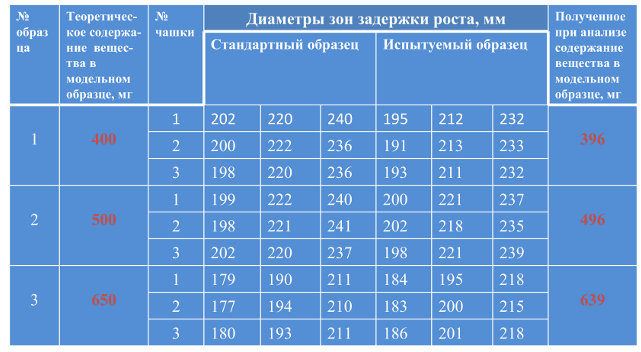
Критерий приемлемости - величина процента восстановления для каждой концентрации: отношение экспериментально полученной концентрации к теоретически рассчитанной (в %)
Процент восстановления должен быть в пределах 98 – 102% • Для образца №1 – 99% • Для образца №2 – 99,2% • Для образца №3 – 98,3% Подтверждена правильность МИ. Подтверждение прецизионности и правильности методики свидетельствуют о ее пригодности для контроля качества данного ЛС или АФИ. Вывод – данная методика гарантирует получение стабильных, достоверных результатов. Валидация методик определения микробиологической чистоты и стерильности Испытание на стерильность – подтверждение отсутствия в испытуемом образце жизнеспособных микроорганизмов Испытание на микробиологическую чистоту: • число бактерий и грибов – не более установленного критерия приемлемости • отдельные виды микроорганизмов не должны обнаруживаться в установленной массе (объеме) ЛС.
Валидация микробиологических методов контроля (микробиологической чистоты и стерильности) - минимально cтатистически подтвержденная пригодность методики, которая проводится на трех различных сериях препарата с учетом всех возможных факторов влияния на достоверность результатов экспериментальных исследований (питательные среды, растворы, персонал).
Поэтому валидационные исследования в отношении определения микробиологической чистоты и стерильности проводят: • на трех различных сериях препарата • трех разных партиях питательных сред • с привлечением трех специалистов, для исследования каждым из них по одной серии препарата • в разные дни (желательно). Методика определения качества лекарственных средств по микробиологическим показателям считывается валидной, если для трех различных серий препарата, на трех разных партиях питательных сред, у трех разных специалистов пригодность методики была подтверждена. Пригодность методики испытания:
1. Использование фармакопейных тест- микроорганизмов и подготовка их в соответствии с фармакопейными требованиями.
2. Подготовка образца - проводится аналогичным образом, как для испытаний микробиологической чистоты нестерильных лекарственных средств или стерильности.
3. Соблюдение условий приготовления питательных сред, проверка их ростовых, индикаторных и ингибирующих свойств (для каждой серии).
4. Проверка наличия/отсутствия антимикробного действия (среднее количество КОЕ на чашках с ЛС должно отличаться не более чем в 2 раза от количества КОЕ в контроле).
5. Нейтрализация антимикробного действия подходящим образом.
6. Предварительная оценка эффективности и безвредности инактиваторов и ПАВ.
7. Проведение контроля (рост культуры на питательной среде без ЛС) и отрицательного контроля (использование для инокуляции стерильного растворителя).
8. Демонстрация возможности обнаружения микроорганизмов- контаминантов в выбранных условиях проведения эксперимента
Валидация методик испытаний: виды и порядок проведения валидации - понятие и виды. Классификация, сущность и особенности.
Оглавление книги открыть закрытьДобавлено: 04 дек 2012 20:46
Хочу поступово викласти частину керівництва ВОЗ, яку я нещодавно переклав на українську.
WHO Expert Committee on Specifications for Pharmaceutical Preparations/ - WHO Technical Report Series, 937, 2006.
Регуляторне керівництво щодо взаємозамінності багатоджерельних (генеричних) лікарських засобів
1. Керівництва щодо реєстраційних вимог для доведення взаємозамінності
Комітет відмітив, що цей документ є переглянутою версією існуючого документу. Комітет затвердив документ в принципі з урахуванням деяких відповідних незначних змін, які стали результатом розгляду коментарів, отриманих на 30.11.2005 (Додаток 7).
3. Список препаратів порівняння для прекваліфікації 1 .
Комітет підтримує документ керівництва, названий «Примітки для заявників щодо вибору препаратів порівняння з точки зору прекваліфікації».
4. Пропозиції щодо відмови від вивчення біоеквівалентності in vivo оральних твердих лікарських форм з негайним вивільненням, включених до Модельного списку життєвонеобхідних лікарських засобів ВООЗ
Комітету був представлений переглянутий документ. Було відмічено, що таблиці необхідно регулярно переглядати, щоб відображати статус Модельного списку життєвонеобхідних лікарських засобів ВООЗ. Незначні поправки були рекомендовані до внесення. Комітет затвердив документ (Додаток 8).
Комітет рекомендував, щоб:
[list:l579lrd.
Добавлено: 19 мар 2012 01:40
Сокращения:
ОТДФ - Оральные Твердые Дозированные Формы
ТДФНВ - Твердые Дозированные Формы Немедленного Высвобождения
ТДФ - Твердые Дозированные Формы
Это руководство разработано для ТДФ немедленного высвобождения (ТДФНВ) и рекомендуется как (1) общие рекомендации для тестирования растворения; (2) подходы разработки спецификаций растворения в соответствии с БСК действующих веществ; (3) статистические методы для сравнения профилей растворения; (4) помощь при определении того, может ли тестирование растворения предоставить вейвер от исследований биоэквивалентности in vivo. Этот документ также дает рекомендации по тестированию растворения, чтобы помочь непрерывно обеспечивать качество препарата и его параметры после каких-то пострегистрационных производственных изменений.обзорная информация по методологии растворения, приборам, условиям проведения тестирования растворения ТДФНВ указана в общем виде в Приложении А. Это руководство должно стать составной частью руководства SUPAC-MR для промышленности: Immediate Release Solid Oral Dosage Forms: Scale-up and Post-Approval Changes: Chemistry, Manufacturing and Controls, In Vitro Dissolution Testing, and In vivo Bioequivalence Documentation с конкретными ссылками на получение профилей растворения для сравнительских целей.
Абсорбция лекарства из ТДФ после орального назначения зависит от высвобождения действующего вещества (ДВ) из препарата, растворения или солюбилизации лекарства при физиологических условиях, и проницания через ЖКТ. Из-за критической природы первых двух стадий растворение in vitro может быть способным предсказать характеристики in vivo. Основываясь на этом общем положении, тесты растворения in vitro для ТДФНВ (капсул, таблеток) используются для (1) оценки качества препарата от серии к серии; (2) планирования разработки новых составов; (3) обеспечения непрерывного качества препарата и его характеристик после некоторых изменений, а именно изменений состава, производственного процесса, места производства, а также масштабирования производственного процесса.
Имеющиеся знания о растворимости, проницаемости, растворении и фармако-кинетике лекарственного средства должны учитываться при определении спецификации для теста растворения при регистрации. Эти знания должна также использоваться для обеспечения постоянной эквивалентности препарата, а также для обеспечения одинаковости препарата при некотором масштабировании и пост-внедренческих изменениях.
Заявки на новые препараты (NDA), подаваемые в FDA, содержат данные по биодоступности и данные по растворению in vitro, которые вместе с данными по химии, производству и контролю (С.
Добавлено: 09 янв 2012 22:54
Руководство для промышленности
Оральные дозированные формы с пролонгированным высвобождением: разработка, оценка и применение корреляций in vitro-in vivo (IVIVC)
FDA, сентябрь 1997
Это руководство дает рекомендации для спонсоров, которые разрабатывают документацию по доказательству корреляции in vitro-in vivo (IVIVC) оральных дозированных форм с пролонгированным высвобождением (ОДФПВ) при подаче документов для лицензирования новой заявки лекарственного средства (NDA), сокращенной заявки нового лекарственного средства (АNDA), или заявки на антибиотик (AADA). Руководство представляет собой всесторонний обзор по (1) методам разработки IVIVC и оценки ее предсказуемости; (2) набору спецификаций по растворению при помощи IVIVC; (3) использованию IVIVC как суррогата биоэквивалентности in vivo при необходимости доказательства биоэквивалентности на стадии первичного лицензирования или в случае возникновения каких-либо до- или пост-регистрационных изменений (например, состава, оборудования, процесса, места производства).
Концепция IVIVC особенно для ОДФПВ активно обсуждалась учеными-фармацевтами. Способность предсказать, точно и правильно, предполагаемые свойства биодоступности для ОДФПВ, исходя из профилей растворения, - сильно желаемая цель. Ряд обзоров и публикаций дают информацию, подтверждающую эту цель.
Эти доклады доказали важность подтверждения IVIVC при оценке параметров биодоступности in vivo для ОДФПВ этому возросла деятельность по IVIVC при подаче заявки на NDA. Однако, еще нет четкого определения полной процедуры разработки IVIVC высокого качества и предсказуемости, а также определения специальной заявки для таких корреляций.
Как часть процедуры, разработанной в этом руководстве, Агентство провело обзоров заявок NDA для лекарственных средств в виде ОДФПВ, чтобы определить время, необходимое для разработки IVIVC. Первый обзор, охватывающий заявки NDA от 1982-92 гг. обнаружил 9 IVIVC в 60 заявках. Более поздний обзор, охватывающий заявки NDA от октября 1994 до октября 1995, обнаружил 9 IVIVC в 12 заявках.
Это руководство основывается на данных предварительных обсуждениях и публикациях, а также текущем понимании FDA и на найденных в другом месте подходах, касающихся разработки реальных и полезных IVIVC. Это руководство излагает уровни корреляций, которые можно получить с разной степенью полезности, важные факторы для данных in vivo и in vitro, оценку корреляции с фокусированием на критические моменты относительно предсказуемости, и практическую пользу, которую можно получить, используя IVIVC. Исходя из этого руководства, спонсоры ободрили.
Добавлено: 26 дек 2011 14:50
Таблетки Глипизид (Glipizide)– биоэквивалентность in vivo и тестирование растворения in vitro
Требования по тестированию in vitro
Тестирование растворения – проводят тестирование растворения на 12 дозированных единицах исследуемого и референтного препаратов, используя биоисследуемые серии. Нужно использовать официально действующий метод растворения по USP (см. Растворение <711>) и следует ссылаться на спонсора. Рекомендуются следующие времена отбора проб для получения профиля растворения:
Времена отбора: 15, 30, 45 и 60 минут.
Тестирование однородности содержания <905> – следует проводить тестирование, как изложено в USP.
Требования по вейверу
Требования по вейверу для исследований по биоэквивалентности in vivo обеспечивается для 5-мг дозировки генерического препарата в соответствии с 21 CFR 320/22(d)(2) при соблюдении следующих условий:Таблетки Глибурид (Glyburide)– биоэквивалентность in vivo и тестирование растворения in vitro
Требования по тестированию in vitro
В данный момент не существует действующей официальной монографии для дозированной формы глибурида в USP.
Тестирование однородности содержания <905> – следует проводить тестирование, как изложено в USP.
Требования по вейверу
Требования по вейверу для исследований по биоэквивалентности in vivo обеспечивается для 1,25- и 2,5-мг дозировок генерического препарата в соответствии с 21 CFR 320/22(d)(2) при соблюдении следующих условий:Добавлено: 26 дек 2011 14:32
Общие правила
Статистические методики для исследований биоэквивалентности при использовании стандартного перекрестного дизайна с двумя последовательностями приема препарата
Введение
Общая методология
Логарифмические превращения фармакокинетических данных
Последовательность влияния
Дальнейшие условия
Оральные дозированные формы пролонгированного высвобождения
Это руководство излагает исследование биоэквивалентности in vivo и тестирования высвобождения in vitro, рекомендуемые для заявок, направляемых для получения торгового разрешения для пролонгированных препаратов, назначаемых орально.
Номенклатура
Дозированные формы с модифицированным высвобождением – такие, у которых характеристики высвобождения ДВ или время высвобождения выбраны так, чтобы добиться терапевтических целей или удобств, которые не могут дать обычные дозированные формы типа растворов, мазей или немедленно растворяющихся дозированных форм. ДФМВ разделяются на два вида: дозированные формы с отсроченным высвобождением и пролонгированным высвобождением. В этом руководстве не рассматривается исследование биоэквивалентности для дозированных форм с отсроченным высвобождением.
Дозированные формы с отсроченным высвобождением – высвобождение ДВ происходит не сразу же после приема.
Дозированные формы с пролонгированным высвобождением – дают, по крайней мере, двукратное уменьшение периодичности дозирования или существенное повышение соблюдения режима лечения или терапевтических свойств по сравнению с обычными дозированными формами (например, раствором или препаратом с немедленным высвобождением, обычно применяемыми твердыми дозированными формами).
Термины контролируемое высвобождение, пролонгированное действие и поддерживающее высвобождение являются синонимами пролонгированного высвобождения. Этот документ использует термин пролонгированное высвобождение, чтобы изложить состав, который не высвобождает ДВ немедленно после орального приема, а также позволяет снизить частоту приемов. Эта номенклатура соответствует полностью определению USP пролонгированного высвобождения, но не специфицирует влияние частоты дозирования. Термины контролируемое высвобождение и пролонгированное высвобождение в этом руководстве рассматриваются как взаимозаменяемые.
Регуляторные указания и общие требования
Исследование биоэквивалентности in vivo для получения торгового разрешения
Исследование биоэквивалентности при помощи однодозовых перекрестных испытаний…
Многодозовые исследования….
Однодозовые перекрестные…







Д. А. Леонтьев, к. фарм.н, заместитель директора по научной работе,
начальник отдела валидации и стандартных образцов
ГП «Украинский научный фармакопейный центр качества лекарственных средств»,
эксперт Европейской фармакопеи,
г. Харьков, Украина
В фармации сложились определенные правила стандартизации, и концепцию неопределенности необходимо использовать с их учетом. Наиболее важными с точки зрения соответствия требованиям GMP являются документы ICH. В них описана терминология валидации; валидационные характеристики, которые изучаются для различных фармацевтических испытаний; даются определенные рекомендации по наполнению эксперимента и т.д. Валидационные характеристики это «инструменты», позволяющие изучить метрологические характеристики методики. Во избежание путаницы приведем английские термины ICH и принятые в ГФУ русскоязычные термины: Accuracy, Trueness «правильность» (степень близости результата к истинному значению); Precision «прецизионность» (степень близости результатов друг к другу). Отметим, что русскоязычный термин «точность» обозначает метрологическую характеристику, которая одновременно характеризует и правильность, и прецизионность. (см. ГОСТ Р ИСО 5725-1-2002 «Точность (правильность и прецизионность) методов и результатов измерений»). Т.е. это величина, обратная неопределенности, и она дает ту же самую информацию, что и неопределенность. Данное понятие в соответствии с идеологией ICH в фармации не используется.
В ICH фактически отсутствуют критерии, которые пользователь вынужден научно обосновать. Весьма примечательна фраза ICH: «Изучение линейности полезно само по себе …». Т.е. ICH рекомендует предоставлять метрологические оценки линейности, но при этом совершенно непонятно, как их оценивать!
Ситуация доходила до крайности: поскольку критерии отсутствовали, любые полученные по формальному формату валидации ICH оценки считались приемлемыми! Как первый шаг стандартизации на предприятии вводились единообразные критерии исходя из здравого смысла: «у нас обычно получается так, значит, мы должны так действовать всегда». Но здравый смысл непонятно как связан с целью сделать риск забраковки выпущенного на рынок препарата приемлемо низким, поэтому «здравый смысл» без научного обоснования может подводить. Таким образом, чтобы эффективно использовать концепцию неопределенности для фармации, необходимо увязать оценки для используемых по формату ICH валидационных характеристик с оценкой неопределенности. Я думаю, что у многих практиков, занимавшихся валидацией при отсутствии научно обоснованных критериев, возникала мысль: «Зачем я занимаюсь этой бессмысленной работой. Ведь что не сделаем все хорошо!».
В ICH недостаточно стандартизован и сам эксперимент по валидации. А от этого зависят требования к валидационным характеристикам. Например, физический смысл коэффициента корреляции характеризует степень «вытянутости» облака наших экспериментальных точек. Если методика остается линейной, то расширение диапазона концентраций (например, от минимального рекомендуемого ICH ±20 до ±50, в процентах от номинального содержания) автоматически приводит к приближению коэффициента корреляции к единице. Таким образом, рекомендации, сделанные с точки зрения «здравого смысла» «хорошо, когда коэффициент корреляции не менее 0.999», на самом деле смысла лишены без указания диапазона, для которого такая рекомендация дается. Аналогичные примеры многочисленны. Таким образом, необходимо увязать требования к валидационным характеристикам с дизайном эксперимента.
НОРМАЛИЗОВАННАЯ СИСТЕМА КООРДИНАТ. Спецификации, концентрации анализируемых растворов и аналитические сигналы для различных веществ могут находиться в самых разных цифровых диапазонах. Например, для дозировок аспирина 100 мг и 500 мг допуски содержания в спецификации будут варьироваться от 95 до 105 мг и от 475 до 525 мг, соответственно. Мы не можем сравнить эти требования, пока не выразим их в процентах от номинального содержания: для обоих препаратов требования ± 5%. Сразу понятно, что и требования к валидационным характеристикам будут одни и те же. Точно так же при проведении валидации: в нашей практике был случай, когда в препарате контролировалось 9 компонентов различными методами анализа и в совершенно разных концентрациях. Если работать в координатах аналитический сигналконцентрация, то для каждого случая необходимо рассчитывать свои критерии приемлемости.
Поэтому в ГФУ (и в Руководстве по валидации) введены нормализованные координаты:

Ci – концентрация анализируемого вещества в i-ом анализируемом растворе (или образце)
Ci st – концентрация этого же вещества в растворе (или образце) сравнения (которая очень близка к номинальной концентрации)
Ai - аналитический сигнал анализируемого вещества для i-ого анализируемого раствора
Аi st - аналитический сигнал этого же вещества для раствора сравнения
Zi – для прямой пропорциональности характеризует значение «найдено»/«введено»
Использование нормализованных координат имеет целый ряд чрезвычайно важных преимуществ:
Появляется унифицированная система критериев, которые определяются только требованиями к максимально допустимой неопределенности результатов анализа maxΔAs. Т.е. для всех ГЛС с допусками содержания ± 5% независимо от дозировок, концентраций анализируемых растворов и от выражения аналитического сигнала (например, для спектрофотометрии оптическая плотность порядка 0.5%; для хроматографии площадь пика порядка миллиона) все критерии будут одинаковыми! Кроме того, все графики (например, для изучения линейности) будут очень наглядными, и не составит труда сравнивать их друг с другом.
Чрезвычайно важно введение величины Z. Z, что позволяет использовать всю совокупность результатов, полученных при изучении линейности (т.е. для разных концентраций), как единый набор «параллельных» измерений. Т.е. из результатов, полученных при изучении линейности, можно также изучать и прецизионность, и правильность методики. Данный момент является критическим, например, для титриметрических методик анализа. При титровании каждая навеска немного отличается от другой, т.е. принципиально отсутствуют параллельные результаты измерений. Другими словами без нормализации результатов по концентрации мы принципиально не можем изучать прецизионность титриметрической методики (да и правильность без усреднения параллельных результатов тоже изучать нельзя!). (Отметим, что в этом виде «найдено»/«введено» можно использовать только в случае прямой пропорциональности, т.е. когда калибровочная прямая проходит через начало координат. Однако это наиболее частый случай в фармацевтическом анализе. В других случаях аналогичным образом вместо Z необходимо использовать значения «найдено»/«введено»).
Еще одно существенное преимущество нормализованных координат связано с природой данных по линейности. Мы имеем дело с физическим законом: аналитический сигнал зависит от концентрации. Например, для спектрофотометрии он называется законом Ламберта-Бугера-Бэра. Задавая концентрацию, мы можем экспериментально изучить, как варьирует оптическая плотность, т.е. оценить метрологические характеристики спектрофотометра (неопределенность его показаний). Для этого существует очень удобный инструмент так называемое остаточное стандартное отклонение s0 это «аналог» стандартного отклонения для параллельных измерений. Но s0 учитывается для результатов, полученных при разных концентрациях. s0 очень легко увязывается с неопределенностью (в данном случае с неопределенностью измерения оптической плотности). Но нас интересует обратная задача: как по оптической плотности найти концентрацию, и какой уровень неопределенности найденного значения концентрации мы получим. Такая обратная зависимость называется калибровочной функцией. Однако мы не можем взять и поменять местами оси X и Y, поскольку по своей природе одна из них является «зависимой» переменной, а другая «независимой». Но когда мы переходим к нормализованным координатам, ситуация принципиально меняется. Для прямой пропорциональности (основной случай для фармацевтического анализа) зависимость, описываемая линейной функцией, проходит через начало координат (или очень близко к ним); угол наклона прямой равен 450 (или очень близок к нему). То есть ордината и абсцисса становятся равнозначными. Поэтому для данного случая можно «перевернуть» оси и использовать s0 для оценки неопределенности калибровочной функции что нас и интересует.
«МЕТРОЛОГИЧЕСКОЕ ЯДРО» ВАЛИДАЦИИ ПО ГФУ. Использование нормализованных координат, линейной модели и принципа незначимости позволило увязать требования ко всем валидационным характеристикам с требованиями к неопределенности. Они базируются на описанном балансе систематических и случайных источников варьирования для различных методов анализа.
Центральная идея валидации по ГФУ использовать результаты из линейности для одновременного изучения правильности и прецизионности. Но это необходимо сделать так, чтобы максимально соответствовать требованиям ICH: для изучения линейности используется не менее 5 концентраций, для изучения правильности не менее 3 концентраций по 3 независимых определения (т.е. в сумме 9). Таким образом, если использовать для изучения линейности 9 различных концентраций, то из этих результатов можно получить оценку правильности и прецизионности в полном соответствии с ICH. Отметим, что рекомендация ICH – использовать для изучения линейности не менее 5 точек годится только для визуальной оценки линейности, но для количественной оценки 5 точек недостаточно.
Кроме этого, все характеристики, получаемые при изучении линейности, также увязаны с оценкой неопределенности это свободный член а, тангенс угла наклона b, остаточное стандартное отклонение s0 и коэффициент корреляции r.
В ГФ СССР и затем в постсоветском пространстве традиционно использовался так называемый линейный коэффициент корреляции r. Его преимущество он позволяет различать как увеличение, так и уменьшение Y при увеличении X. Однако в фармацевтическом анализе для оценки линейности калибровочной функции знак наклона не имеет значения. Его недостаток сложное математическое выражение. Это привело к тому, что USP в проекте общей статьи <1220> «Статистические инструменты валидации аналитических методик» (Pharmacopoeal Forum 40(5)) высказала мнение, что коэффициент корреляции не может быть увязан с оценкой неопределенности.
Поэтому в ГФУ используется так называемый общий индекс корреляции RC (частным случаем которого является линейный коэффициент корреляции). Ограничение RC – он изменяется от 0 до 1, т.е. не различает увеличения и уменьшения Y с ростом X. Однако в силу своей простоты и наглядности RC может быть увязан с требованиями к неопределенности результата анализа. При небольших значениях степеней свободы (f) r дает некорректные результаты, которые очень мало отличаются от соответствующих значений, рассчитанных для RC. Однако при f ≥ 5 значения r и RC практически совпадают. Далее под обозначением r понимается именно RC.
Валидационные характеристики оценивают систематические или случайные источники варьирования и фактически могут быть сведены к характеристике Правильность (оценка систематического варьирования) или Прецизионность (оценка случайного варьирования).
Таким образом, даже на первый взгляд такие «качественные» валидационные характеристики, как Специфичность и Робастность, могут и должны оцениваться количественно.
Значимые источники варьирования (и систематические, и случайные) могут иметь различные (и вполне объяснимые) причины. Например, если неудачно выбран диапазон концентраций и калибровка при высоких концентрациях начинает «загибаться», то пострадают валидационные характеристики Правильность, Прецизионность, Линейность свободный член а, s0 и r. Если недостаточна Специфичность методики пострадают валидационные характеристики Правильность, Линейность свободный член а. Если плохо подобраны температура и время выдержки образца в парофазном анализе, можно получить очень плохую Прецизионность и т.д. Для случайного варьирования мы не знаем, в какую сторону отклонится следующий результат, но мы можем влиять на амплитуду этого варьирования.
Наиболее общий рецепт оценки неопределенности это оценивать суммарный вклад для всех источников варьирования («сводить баланс»). Однако мы не знаем причины, которая вызвала завышение свободного члена или ухудшение Правильности и т.п. (если мы знаем причину, мы можем ее устранить и сделать данный источник варьирования незначимым!). Поэтому попытка сведения баланса приводит нас к неразрешимой проблеме чтобы не учесть в оценке неопределенности несколько раз один и тот же источник варьирования, мы должны заранее знать его природу. Но почему тогда мы не оптимизировали методику?!
Решить данную проблему позволяет использование принципа незначимости. Для хорошо разработанной хроматографической или спектрофотометрической методики все систематические источники варьирования должны быть незначимы. Тогда вся максимально допустимая неопределенность отводится на случайные источники варьирования, причину которых мы не ищем. (Если нужно, мы ужесточаем требования к случайному варьированию с учетом вклада от пробоподготовки, которая является отдельным источником варьирования). Данный подход очень хорошо себя зарекомендовал при практическом проведении валидации и отражен в таблице ниже. (Для титрования наблюдается обратная ситуация для хорошо разработанной методики должны быть незначимы случайные источники варьирования).
*В допуски содержания для количественного определения; δ – систематическая составляющая варьирования (правильность); КО – Количественное определение; ОС Однородность содержания для дозированных лекарственных форм; Р Растворение для твердых дозированных лекарственных форм.
Предел обнаружения (ПО) и предел количественного определения (ПКО) в ГФУ также увязаны с требованиями к неопределенности. Если ПО/ПКО находится слишком близко к предельно допустимой концентрации примеси, то мы можем произвольно то браковать препарат (мы «видим» примесь), то «подтверждать качество» («не видим» примесь). Надежность принятия корректного заключения о качестве 95% обеспечивается, если ПО/ПКО составляет не более 0.32 от предельно допустимой концентрации.
СТАНДАРТИЗОВАННЫЕ ПРОЦЕДУРЫ ПРОВЕДЕНИЯ ВАЛИДАЦИИ. Проведение валидации имеет свою специфику для различных методов анализа (иногда это принципиальные различия например, титрование и сравнительные методы анализа). В связи с этим были разработаны СТАНДАРТИЗОВАННЫЕ ПРОЦЕДУРЫ ПРОВЕДЕНИЯ ВАЛИДАЦИИ для конкретных методов анализа. Все эти процедуры опираются на изложенную выше концепцию неопределенности. Поскольку постановка эксперимента для изучения валидационных характеристик может варьировать, предложенная методология проведения эксперимента не внесена в ГФУ. Данные процедуры опубликованы, и ими можно пользоваться без какого-либо дополнительного научного обоснования достаточно ссылки на соответствующую публикацию:
Количественное определение методом спектрофотометрии (Фармаком 2004 №3).
Количественное определение хроматографическими методами (Фармаком 2010 №1).
Количественное определение методами титриметрии (Фармаком 2009 №2).
Определение родственных примесей приборными хроматографическими методами (Фармаком 2005 №2/3).
Определение остаточных органических растворителей методом газовой хроматографии (Фармаком 2005 №4).
Количественное определение методом атомно-абсорбционной спектрофотометрии (Фармаком 2011 №4).
Количественное определение методом спектрофотометрии в варианте удельного показателя поглощения (Фармаком 2004 №12).
Количественное определение для суммарных препаратов биологического происхождения методом калибровочного графика (Фармаком 2013 № 3).
Исследование биоэквивалентности in vitro по профилям растворения для твердых дозированных лекарственных форм (Фармаком 2006 №4; 2013 № 4).
Заключение. Подход, описанный в ГФУ (и в Руководстве по валидации под редакцией Н.В. Юргеля), является на настоящий момент единственным, в котором используется формат проведения валидации ICH, и при этом все валидационные характеристики используются как инструменты для оценки неопределенности. Отметим, что USP в проекте общей статьи <1220> сделала попытку оценивать из результатов валидации неопределенность результатов анализа, однако предложенный формат проведения валидации несовместим с форматом ICH и даже с форматом общей статьи USP <1225> «Валидация аналитических методик». Данная общая статья до сих пор не введена в USP.
Оценка неопределенности делает процедуру валидации «осмысленной», т.к. дает Производителю чрезвычайно важную информацию достаточно ли низкий риск забраковки препарата при его обращении по причине некачественной методики.
Разработаны все метрологические «инструменты», позволяющие практически проводить валидацию в данном формате. Разработан уникальный инструмент прогноз неопределенности, который опирается на максимально допустимую неопределенность для частных составляющих неопределенности, которая принята в фармацевтической отрасли. Это чрезвычайно действенный инструмент, т.к. позволяет оценить очень важные аспекты метрологической корректность методики уже исходя из текста методики.
Это чрезвычайно важно, т.к. очень часто в руководствах (и GMP, и ISO) задекларированы намерения «нужно контролировать неопределенность», при этом все практические вопросы должен решить пользователь. Это привело, например, к тому, что лаборатории OMCL для валидированных методик вообще не оценивают их метрологическую корректность, только если возникает проблема с забраковкой препарата. Однако опыт показывает, что «в наследство» от валидации, не опирающейся на научно обоснованные критерии, нам досталась масса совершенно очевидно некорректных методик (с точки зрения концепции неопределенности, которая опирается на предназначение фармацевтических испытаний), которые включены и в ведущие фармакопеи. Т.е. перед лабораториями была поставлена непосильная задача и они «вернули мяч» фактически отказались от оценки неопределенности.
Для выполнения расчетов, описанных в ГФУ («метрологическое ядро»), ГП «Фармакопейный центр» разработано программное обеспечение, позволяющее автоматизировать работу и существенно снизить риск ошибки при оформлении результатов.
Разработанный подход легко переносится на связанные с валидаций процедуры Верификация методик и их Передача (Analytical Procedure Transfer), которые упоминаются как обязательные в новой редакции GMP PIC/S.
01 сентября 2015
Elena