
_p0_Cover.jpg)



_p1_Titul_min.png)




 Рейтинг: 4.3/5.0 (1866 проголосовавших)
Рейтинг: 4.3/5.0 (1866 проголосовавших)Категория: Руководства
Стремление некоторых кругов и даже государств монополизировать достижения современной генетики, в том числе информацию, полученную в результате расшифровки генома человека, рассматривается всеми честными исследователями и специалистами как неправомерное и не подлежащее удовлетворению. Расшифрованный геном человека должен быть поставлен на благо всего человечества. Как считает ЮНЕСКО, «геном человека лежит в основе изначальной общности всех представителей человеческого рода, а также признания их неотъемлемого достоинства и разнообразия. Геном человека знаменует собой достояние всего человечества». Эта позиция ЮНЕСКО должна быть поддержана всем гражданским обществом и лечь в основу формирования морально-нравственных принципов человечества.
Генетический скрининг, если он проводится с целью устранения тяжелых наследственных заболеваний и производится на основе добровольного информированного согласия, несет в себе благоприятные возможности для отдельного человека и для общества. Поэтому он не противоречит современной концепции биомедицинской этики и может развиваться.
Современные генно-инженерные технологии еще не достаточно отработаны и таят в себе ряд опасностей. В будущем генная
терапия сможет излечить пациента от ряда наследуемых болезней, поэтому ее следует считать приемлемой и отвечающей современной концепции биомедицинской этики.
Морально-нравственные основы фетальной терапии должны основываться на информированном согласии женщин на использование абортивного материала и информировании ее о предполагаемых манипуляциях и об использовании извлеченных тканей. Взятие абортивного материала для косметических целей большинство гражданского общества считает недопустимым.
Генетическая паспортизация должна быть, в основном, ориентирована на медицинские задачи, при этом такая информация должна быть защищена во избежание злоупотребления ею. Процесс сбора информации о генах населения Земли должен быть поставлен под строгий этический контроль.
Генетическая модификация продуктов является перспективной областью для получения продуктов питания и лекарственных препаратов. Но и это направление требует выработки и совершенствования морально-нравственных позиций, соответствующих состоянию общества, а также контроля, включающего такой важный элемент, как непреднамеренное опыление традиционных растений пыльцой генно-инженерных видов, что может привести к непредвиденным последствиям.
Таким образом, современные достижения генетики, в том числе генная инженерия, представляются весьма перспективной областью для фармации. К сожалению, на вузовском этапе системы непрерывного фармацевтического образования эти проблемы пока изучаются недостаточно.
НОРМАТИВНО-ПРАВОВОЕ РЕГУЛИРОВАНИЕ И ЭТИЧЕСКИЕ
ПРОБЛЕМЫ В СИСТЕМЕ ДОКЛИНИЧЕСКИХ И КЛИНИЧЕСКИХ
4.1. Доклинические исследования лекарственных средств
Целью доклинических исследований лекарственных средств является получение научными методами оценок и доказательств их эффективности и безопасности.
Доклинические исследования лекарственных средств включают в себя химические, физические, биологические, микробиологические, фармакологические, токсикологические и другие экспериментальные исследования. Все они взаимозависимы. Так, в результате изучения острой токсичности потенциальных лекарственных соединений получают данные для последующих фармакологических исследований, которые, в свою очередь, определяют степень и продолжительность изучения хронической токсичности вещества.
При фармакологических исследованиях определяют терапевтическую эффективность препарата, а также его влияние на основные анатомические и физиологические системы организма. В процессе изучения фармакодинамики вещества устанавливают не только его специфическую активность, но и возможные побочные реакции, связанные с фармакологической активностью. Действие исследуемого препарата на здоровый и больной организм может различаться, поэтому фармакологические испытания должны проводиться на моделях соответствующих заболеваний или патологических состояний.
При токсикологических исследованиях устанавливают характер и выраженность возможного повреждающего воздействия препаратов на экспериментальных животных. В токсикологических исследованиях выделяют три этапа:
1) изучение острой токсичности вещества при однократном введении;
2) определение хронической токсичности соединения, которое включает в себя повторные введения препарата на протяжении одного года, а иногда и более;
3) установление специфической токсичности препарата — онкогенности, мутагенности, эмбриотоксичности, включая тератогенное
действие, аллергизирующие свойства, а также способности вызывать лекарственную зависимость.
Доклинические исследования лекарственных средств в России проводятся в соответствии с Федеральным законом «О лекарственных средствах». Согласно статье 36 закона «Доклинические исследования лекарственных средств» доклинические исследования проводятся организациями-разработчиками лекарственных средств по правилам лабораторной практики, утвержденным федеральным органом контроля качества лекарственных средств. Доклинические исследования на животных проводятся в соответствии с международными правилами. Контроль за соблюдением правовых и этических норм использования животных при проведении доклинических исследований лекарственных средств осуществляется соответственно федеральным органом контроля качества лекарственных средств и территориальными органами.
Правила лабораторной практики в Российской Федерации (GoodLaboratoryPractice—GLPРФ) (утверждены Приказом Министерства здравоохранения Российской Федерации от 19.06.2003 № 267) — это процедуры и требования в отношении оценок лекарственных средств и использования экспериментальных животных при этом. Данным документом введено обязательное составление и утверждение протокола эксперимента, одобрение его этическим комитетом или комиссией для определения допустимости и приемлемости эксперимента. Также введена необходимость разработки и соблюдения стандартных операционных процедур для всех стадий и операций по обращению с животными. Исследователи должны обладать соответствующей квалификацией и опытом, т.к. несут они ответственность за анализ результатов и представление отчета о своей работе руководителю испытания.
В документе указано, что доклинические исследования лекарственных средств на животных проводятся в соответствии с международными правилами, с максимальной гуманностью умерщвления животных и проведения самих экспериментов. Потребление животными пищи и воды происходит в соответствии с протоколом исследований и принципом отсутствия в них патогенных микроорганизмов и вредных примесей. Должно достигаться максимальное документирование всех действий по приему, обслуживанию и экспериментированию с животными. Большое внимание уделяется стандартизации тест-систем, то есть использованию для экспериментов барьерных животных, живущих в стерильных условиях, лишенных
патогенной микрофлоры, с определенным генетическим статусом. Это дает возможность сократить разброс результатов, само количество экспериментов и, соответственно, количество участвующих в них животных. Так, если необходимо получить высокостандартные и воспроизводимые результаты, в опытах применяют линейных (гомозиготных) животных, которых получают путем многократного близкородственного скрещивания. Такие животные характеризуются определенными биологическими свойствами, например, восприимчивостью к инфекционным агентам, способностью к иммунному ответу.
GLPРФ определяют необходимость достаточного количества помещений для животных или площадей для обеспечения раздельного содержания видов; животные должны содержаться при соответствующих условиях окружающей среды и в помещениях, подвергаемых уборке и удалению отходов. При проведении экспериментов обязательны правила обезболивания и эвтаназии. Содержание экспериментальных животных должно соответствовать действующим Санитарным правилам по устройству, оборудованию и содержанию экспериментально-биологических клиник (вивариев).
Необходимость проведения доклинических исследований лекарственных средств обусловлена следующим:
• проведенные исследования фармакологических средств на животных в соответствии с современными требованиями являются основой для оценки их безопасного использования в медицинской практике;
• эксперименты на животных необходимы для развития медико-биологических наук, поскольку позволяют лучше понимать законы и механизмы жизненных процессов;
• изучение повреждающего действия исследуемого препарата на организм экспериментальных животных позволяет определить, какие органы и ткани наиболее чувствительны к данному веществу и на что следует обратить особое внимание при клинических испытаниях. Только в опытах на животных можно выявить воздействие изучаемого вещества на органы с использованием гистологических методов оценки их структуры, влияние препаратов на внутриутробное развитие плода, возможное мутагенное или канцерогенное действие и ряд других воздействий;
• «Чем полнее будет проделан опыт на животных, тем менее часто больным придется быть в положении опытного объекта со всеми печальными последствиями» (И.П. Павлов).
Обоснование необходимости предварительного проведения опытов на животных перед первым испытанием новых лекарств на людях содержится в Хельсинкской декларации, где утверждается, что биомедицинские исследования на людях должны отвечать общепринятым научным принципам и основываться на адекватно выполненных лабораторных опытах и экспериментах на животных, а также исчерпывающем знании научной литературы. Кроме того, этот кодекс этических принципов призывает к бережному отношению к животным, которых используют для исследований.
Требование этичности стало обязательным условием проведения экспериментов на животных во всех странах мира. Это показатель цивилизованности страны. В начале 1985 г. Совет международных медицинских научных организаций (СММНО) опубликовал «Этический кодекс», который содержит «международные рекомендации по проведению медико-биологических исследований с использованием животных». В этом кодексе сформулированы приемлемые для научных работников и для общественных групп защитников животных теоретические принципы и этические правила, которые могут быть приняты за основу при разработке регламентирующих мер и нормативных документов в разных странах мира в отношении использования животных для биомедицинских исследований. Рекомендации составлены, исходя из следующих положений:
• в принципе использование животных для научных целей нежелательно;
• по возможности следует применять методы, не требующие использования животных;
• при существующем уровне знаний использование животных является неизбежным;
• моральный долг ученых — гуманно относиться к подопытным животным, по возможности не причинять им боли и неудобств и постоянно стремиться изыскивать способы получения того же результата без привлечения живых животных;
• животным, предназначенным для медико-биологических исследований, следует обеспечивать наилучшие из возможных условия их содержания.
В последние десятилетия для решения этических проблем при использовании животных в экспериментальной биологии и медицине руководствуются концепцией «трех R» Рассела и Берча: замена (replacement), уменьшение (reduction), повышение качества
(refinement). Такой подход к экспериментированию имеет своей целью применение лучших научных методов при одновременном сокращении количества животных, используемых в экспериментах, а также при усовершенствовании экспериментальной техники с целью минимизации страданий, испытываемых подопытными.
Во многих странах, в том числе в странах-участниках Европейского Союза, закон обязывает исследователей, подающих заявление о лицензии на проект, объявить, что они полностью учли возможность применения альтернативных методов экспериментирования. Это поддерживается Директивой ЕС 86/609/ЕЕС, а Соглашение ЕС по защите позвоночных животных (1986) постановляет: проведение эксперимента запрещается, если имеется другой научно приемлемый метод получения желаемого результата — без использования животных.
К альтернативным методам относятся следующие:
• улучшенная система хранения и использования информации, а также обмен информацией об экспериментах, уже проведенных над животными, во избежание повторения таких процедур;
• использование физических и химических приемов и прогнозов, основанных на физических и химических свойствах молекул;
• использование математических и компьютерных моделей, в том числе моделирование количественных отношений типа «структура-деятельность»; молекулярное моделирование и использование компьютерных графических средств; моделирование биохимических, физиологических, фармакологических, токсикологических и поведенческих систем и процессов;
• использование invitro-методов, в том числе подклеточных фракций, кратковременного хранения слоев ткани, суспензии клеток и обрызгивания органов, а также выращивания тканей (клеточное и органотипичное), в том числе выращивания человеческих тканей;
• использование низших организмов с ограниченной чувствительностью и/или не защищенных законодательством, регулирующим эксперименты над животными;
• использование позвоночных животных на ранних этапах их развития, предшествующих той стадии, на которой регулируется их использование в экспериментах и других научных процедурах;
• эксперименты с участием людей, включая участников-добровольцев, после маркетинговых эпидемиологических наблюдений.
Внедрение альтернативных методов позволит сократить число животных, применяемых для получения информации; уменьшить частоту или интенсивность негуманных процедур; заменить животных на альтернативные биологические модели (АБМ). Уникальными альтернативными биологическими моделями признаны культуральные — органы, ткани, клетки вне организма, «invitro». Ими могут быть бактерии, оплодотворенные куриные яйцеклетки, эмбрионы лягушек, инфузории и пр. АБМ обеспечивают высокую степень воспроизводимости, следовательно, и статистическую достоверность получаемых результатов.
Согласно современным представлениям, все шире распространяется мнение о трудности определения безопасности (или опасности) для человека лечебного или профилактического средства на основании доклинических испытаний, проведенных только на животных.
Насколько низка эффективность использования животных в тестировании лекарственных препаратов, показывает тот факт, что примерно 90% новых лекарственных средств забраковываются на ранних стадиях клинических испытаний, хотя они прошли многолетние испытания на животных по специальной схеме, включающей тесты на острую и хроническую токсичность, канцерогенность, мутагенность и тератогенность.
Известно много случаев, когда опыты на животных привели к неправильным выводам о безопасности препарата. Большой резонанс в мире получило так называемое «дело о талидомиде», успокаивающем средстве, которое прошло испытание на животных и было рекомендовано для приема беременным женщинам.
Причины низкой эффективности экспериментов на животных определяются биологическими различиями между человеком и экспериментальными животными. Животные могут по-разному реагировать на один и тот же лекарственный препарат, это обусловлено следствием различий в абсорбции, в кишечной флоре, в распределении в тканях, в метаболизме, включая биоинтоксикацию и детоксикацию, в механизмах и скорости всасывания, распределения метаболизма лекарственных веществ. Все это делает проблематичной экстраполяцию результатов, полученных на животных, на человека.
Большинство болезней человека возникает и протекает совершенно отлично от болезней животных, даже если они имеют одно и то же название. Возникновение травм, влияние патогенных и токсичных повреждений на человека существенно отличается от
таковых у животных. Организм человека сложнее организма любого животного, его связи с окружающей средой отличаются от всех форм связей у животных.
Научно-технический прогресс «создал» новые болезни, которых не было в древнюю эпоху жизни человека. Возникли радиационные, авиационные, автомобильные болезни, обширная группа профессиональных заболеваний, которые вызываются особенностями трудового процесса или вредными воздействиями материалов производства. Социальные факторы создают у людей особые, свойственные преимущественно человеку болезни, не встречающиеся у животных. Существенным различием патологических процессов у людей и животных является значительно большее разнообразие форм и вариантов болезней у человека. Поэтому, в лучшем случае, у животных воспроизводятся только отдельные элементы болезней человека, некоторые их «составные части» — симптомы и синдромы, но не заболевание в целом. По существу, ни одно инфекционное заболевание человека не протекает у животных, восприимчивых к конкретному микроорганизму, тождественно с его клиническим течением и развитием у людей.
Учитывая данные факты, не нужно забывать и о положительных результатах, которые дают эксперименты на животных для развития медицины и фармации и для всего человечества. Поэтому на исследователей ложится большой груз ответственности при обосновании необходимости проведения доклинических исследований, соблюдения законодательных и этических норм при проведении экспериментов на животных, при интерпретации итогов исследования. Таким образом, от высокого нравственного уровня исследователя будет зависеть гарантия надежности результатов, доказывающая целесообразность, осмысленность участия животных в эксперименте.
4.2. Клиническое исследование лекарственных средств
Клиническое исследование (КИ) — это изучение клинических, фармакологических, фармакодинамических свойств исследуемого препарата у человека, включая процессы всасывания, распределения, изменения и выведения, с целью получения научными методами оценок и доказательств эффективности и безопасности лекарственных средств, данных об ожидаемых побочных явлениях и эффектах взаимодействия с другими лекарственными средствами.
Целью КИ лекарственных средств является получение научными методами оценок и доказательств эффективности и безопасности лекарственных средств, данных об ожидаемых побочных эффектах от применения лекарственных средств и эффектах взаимодействия с другими лекарственными средствами.
В процессе КИ новых фармакологических средств выделяют 4 взаимосвязанные фазы:
1. Определяют безопасность лекарственных средств и устанавливают диапазон переносимых доз. Исследование проводят на здоровых мужчинах-добровольцах, в исключительных случаях — на больных.
2. Определяют эффективность и переносимость лекарственных средств. Подбирается минимальная эффективная доза, определяются широта терапевтического действия и поддерживающая доза. Исследование проводят на больных той нозологии, для которой предназначен исследуемый препарат (50-300 лиц).
3. Уточняют эффективность и безопасность препарата, его взаимодействие с другими лекарственными средствами в сравнении со стандартными методами лечения. Исследование проводят на большом числе пациентов (тысячи больных), с привлечением особых групп больных.
4. Пострегистрационные маркетинговые исследования изучают токсические действия препарата при длительном приеме, выявляют редкие побочные эффекты. В исследование могут быть включены разные группы больных — по возрасту, по новым показаниям.
Виды клинических исследований:
• открытое, когда все участники испытаний знают, какой препарат получает больной;
• простое «слепое» — больной не знает, а исследователь знает, какое лечение было назначено;
• в двойном «слепом» — ни штат исследователей, ни больной не знают, получает ли он препарат или плацебо;
• тройное «слепое» — ни штат исследователей, ни проверяющий, ни больной не знают, каким препаратом он лечится.
Одной из разновидностей КИ являются исследования на биоэквивалентность. Это основной вид контроля воспроизведенных лекарственных средств, не отличающихся лекарственной формой и содержанием действующих веществ от соответствующих оригиналов. Исследования биоэквивалентности позволяют сделать обоснованные
заключения о качестве сравниваемых препаратов по меньшему объему первичной информации и в более сжатые сроки. Проводятся в основном на здоровых добровольцах.
На территории России осуществляются клинические исследования всех фаз. Большая часть международных клинических исследований и исследований зарубежных лекарственных средств относится к 3-й фазе, а в случае клинических исследований отечественных лекарственных средств значительную их часть составляют исследования 4-й фазы.
В России за последние десять лет сложился специализированный рынок клинических исследований. Он хорошо структурирован, здесь работают высококвалифицированные профессионалы — врачи-исследователи, научные работники, организаторы, менеджеры и др. активно действуют предприятия, строящие свой бизнес на организационных, сервисных, аналитических аспектах проведения КИ, среди них — контрактно-исследовательские организации, центры медицинской статистики.
За период с октября 1998 г. по 1 января 2005 г. были поданы документы с просьбой разрешить 1840 клинических исследований. В 1998-1999 гг. отечественные компании составляли крайне незначительную долю заявителей, но начиная с 2000 г. их роль заметно возросла: в 2001 г. было 42%, в 2002 г. — уже 63% заявителей, в 2003 г. — 45,5%. Среди зарубежных стран-заявителей первенствуют Швейцария, США, Бельгия, Великобритания.
Объектом изучения клинических исследований являются лекарственные средства как отечественного, так и зарубежного производства, область применения которых затрагивает практически все известные разделы медицины. Наибольшее количество лекарственных средств применяется для лечения сердечно-сосудистых и онкологических заболеваний. Далее следуют такие области, как психиатрия и неврология, гастроэнтерология, инфекционные болезни.
Одной из тенденций в развитии сектора клинических испытаний в нашей стране следует признать быстрый рост числа КИ на биоэквивалентность препаратов-дженериков. Очевидно, что это вполне соответствует особенностям российского фармацевтического рынка: как известно, он представляет собой рынок воспроизведенных препаратов.
Проведение клинических испытаний в России регулируетсяКонституцией Российской Федерации, которая гласит, что «. никто не
может быть без добровольного согласия подвергнут медицинским, научным и иным опытам».
Некоторые статьи Федерального закона «Основы законодательства РФ об охране здоровья граждан» (от 22.07.1993, № 5487-1) определяют основы проведения КИ. Так, в статье 43 указано, что не разрешенные к применению, но находящиеся на рассмотрении в установленном порядке лекарственные средства, могут использоваться в интересах излечения пациента только после получения его добровольного письменного согласия.
Федеральный Закон «О лекарственных средствах» № 86-ФЗ имеет отдельную главу IX «Разработка, доклинические и клинические исследования лекарственных средств» (статьи 37-41). Здесь указан порядок принятия решения о проведении КИ ЛС, правовые основы проведения КИ и вопросы финансирования КИ, порядок их проведения, права пациентов, участвующих в КИ.
Клинические исследования проводятся в соответствии со Стандартом отрасли ОСТ 42-511-99«Правила проведения качественных клинических испытаний в Российской Федерации» (утверждено Минздравом России 29 декабря 1998 г.) (Good Clinical Practice — GCP). Правила проведения качественных клинических испытаний в Российской Федерации представляют собой этический и научный стандарт качества планирования и проведения исследований на человеке, а также документального оформления и представления их результатов. Соблюдение этих правил служит гарантией достоверности результатов клинических испытаний, безопасности, охраны прав и здоровья испытуемых в соответствии с основополагающими принципами Хельсинкской декларации. Требования данных Правил должны соблюдаться при проведении клинических испытаний лекарственных средств, результаты которых планируется представить в разрешительные инстанции.
GCPустанавливают требования к планированию, проведению, документальному оформлению и контролю клинических исследований, призванных гарантировать защиту прав, безопасность и охрану здоровья лиц, в них участвующих, при проведении которых нельзя исключить нежелательного влияния на безопасность и здоровье человека, а также обеспечить достоверность и точность получаемой в ходе исследования информации. Правила обязательны для выполнения всеми участниками клинических исследований лекарственных средств на территории Российской Федерации.
В целях совершенствования методических основ проведения исследований биоэквивалентности лекарственных средств, являющихся основным видом медико-биологического контроля воспроизведенных лекарственных средств, Министерством здравоохранения и социального развития Российской Федерации 10.08.2004 г. утверждены методические указания «Проведение качественных клинических исследований биоэквивалентности лекарственных средств».
Согласно нормативным документам, КИ ЛС проводятся в учреждениях здравоохранения, аккредитованных федеральным органом исполнительной власти, в компетенцию которого входит осуществление государственного контроля и надзора в сфере обращения лекарственных средств; он же составляет и публикует перечень учреждений здравоохранения, имеющих право проводить клинические исследования лекарственных средств.
Правовую основу проведения КИ ЛС составляют решение федерального органа исполнительной власти, в компетенцию которого входит осуществление государственного контроля и надзора в сфере обращения лекарственных средств, о проведении КИ лекарственного средства и договор о его проведении. Решение о проведении КИ ЛС принимается Федеральной службой по надзору в сфере здравоохранения и социального развития Российской Федерации в соответствии с законом «О лекарственных средствах» и на основании заявления, положительного заключения комитета по этике при федеральном органе контроля качества лекарственных средств, отчета и заключения о доклинических исследованиях и инструкции по медицинскому применению лекарственного средства.
При федеральном органе контроля качества лекарственных средств создан Комитет по этике. Учреждение здравоохранения не начинает исследование, пока Комитет по этике не одобрит (в письменном виде) форму письменного информированного согласия и другие материалы, предоставляемые испытуемому или его законному представителю. Форма информированного согласия и другие материалы могут пересматриваться по ходу исследования, если открываются обстоятельства, способные повлиять на согласие испытуемого. Новая редакция перечисленной выше документации в обязательном порядке проходит одобрение Комитета по этике, а факт доведения ее до испытуемого должен быть документально подтвержден.





















«Чем полнее будет проделан опыт на животных, тем менее часто больным придется быть в положении опытного объекта со всеми печальными последствиями»

Доклинические исследования - биологические, микробиологические, иммунологические, токсикологические, фармакологические, физические, химические и другие исследования фармакологического средства путем применения научных методов оценок в целях получения доказательств безопасности, качества и эффективности фармакологического средства.
По данным американской Ассоциации разработчиков и производителей лекарственных препаратов (PhRMA) из 10 тысяч лекарств-кандидатов, взятых фармацевтическими компаниями в разработку, на стадию доклинических исследований выходит 250 (или 2,5 %), при этом на стадию клинических исследований попадают 5 (или 0,05 %) и только один из кандидатов становится лекарственным препаратом и поступает в широкую медицинскую практику. Весь этот процесс занимает в среднем 10-12 лет. Как видно, из представленных данных, на стадии доклинических исследований происходит значительная селекция лекарств-кандидатов, обусловленная обширными исследованиями в тестах in vitro (лабораторные исследования в пробирках) и in vivo (исследования на лабораторных животных), в ходе которых получают предварительные данные о токсичности, эффективности, фармакологических свойствах, фармакокинетике и метаболизме изучаемого лекарственного препарата. Данный блок работы трудоемкий, финансовозатратный и очень важный.
В настоящее время арсенал методов доклинической оценки фармакологической активности и токсичности лекарственного препарата значительно расширен.
Доклинические исследования начинаются с момента, когда синтезирована новая потенциально эффективная молекула лекарственного средства, предварительно пройдя селекцию из множества потенциальных молекул. В тех случаях, когда у новой молекулы выявляется определенная биологическая активность, ее подвергают тщательным доклиническим исследованиям, которые проводят по стандартным унифицированным методикам.
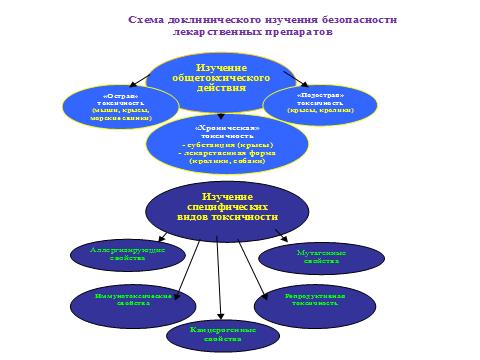
Доклинические исследования безопасности лекарственных препаратов выявляют потенциально повреждающее действие лекарственного препарата и оценивают безопасность его применения. Изучают общетоксическое действие препарата (острая, подострая/хроническая токсичность, местно-раздражающее действие), специфическую токсичность: его влияние на половое поведение и на репродуктивные функции самцов и самок, проводят оценку тератогенной опасности, перинатальных и постнатальных эффектов, лактации, аллергизирующих, иммунотоксических свойств.
Во время проведения токсикологического исследования наши специалисты определяют, можно ли использовать лекарственный препарат в клинической практике, также они тщательно вычисляют допустимые дозы потребления лекарственного препарата. Во время токсикологического анализа выявляются побочные эффекты, и устанавливается запрет на применение лекарства. Вся работа над токсикологическим исследованием проводится в специально оборудованной лаборатории, на животных прошедших необходимую карантинизацию.
Доклинические исследования эффективности лекарственных препаратов — оценка фармакологической активности лекарства-кандидата для лечения, профилактики, диагностики того или иного заболевания, а так же фармакодинамических свойств ЛС. Для изучения специфической фармакологической активности лекарственного средства проводят доклиническое исследование на моделях заболеваний/синдромов у лабораторных животных.
Для выбора доз и схем применения лекарственного препарата проводят фармакокинетические доклинические исследования, включающие исследования всасываемости, распределения, метаболизма и выведения препарата из организма. Данные доклинические исследования также являются незаменимым фрагментом оценки биоэквивалентности дженерикового лекарственного препарата. Фармакокинетическое доклиническое исследование важно также и во время испытания нового лекарственного препарата, которое содержит одно действующее вещество или для комплексного препарата, у которого выражено действующее вещество. Выводы фармакокинетического исследования применяют при утверждении схемы дозировки разрабатываемого лекарственного препарата, способа его использования в клинической практике.
Доклинические исследования позволяют оценить токсичность препарата, установить зависимость между дозой вводимого препарата и эффектом, определить наиболее чувствительные к препарату органы и системы, изучить эффекты, которые невозможно или сложно исследовать у человека, и с определенной долей вероятности прогнозировать степень безопасности его применения в клинике и предсказать возможное побочное действие. По мнению специалистов ВОЗ, экстраполяция данных с животных на человека возможна лишь при использовании многих видов животных, а также одинаковая реакция на исследуемый препарат у 3-4 видов животных обеспечивает совпадение с клиническими данными примерно в 70 % случаев.
Компания «СиТиЭр Фарма» предлагает полный спектр услуг по планированию и организации доклинических исследований как оригинальных, так и воспроизведенных лекарственных средств и фармацевтических субстанций:
Доклинические исследования проводятся в соответствие с российскими и международными требованиями на ведущих российских лабораторных базах с соблюдением правил GLP:
Обратившись в нашу компанию, вы не только получите качественно проведённое доклиническое исследование, но и получите скидку на работу при проведении полного блока исследования.
Объем доклининческих исследований
Результаты доклинических исследований лекарственных средств необходимо представить с целью регистрации или проведения клинических исследований лекарственного препарата в РФ (№ 61-ФЗ от 12 апреля 2010 г. статья 18, часть 3, пункт 9 и статья 20, часть 1).
Необходимый объем доклинических исследований:
1. Воспроизведенные лекарственные средства:
— острая и подострая (субхроническая) токсичность, местнораздражающее действие в сравнении с зарегистрированным аналогом.
2. Оригинальные лекарственные средства:
2.1. Общетоксические свойства:
острая и подострая (субхроническая) токсичность, хроническая токсичность, местнораздражающее действие.
2.2. Специфические виды токсичности:
— репродуктивная токсичность;
— канцерогенное действие;
— аллергизирующее действие;
— иммунотоксическое действие.
2.3. Фармакологическая безопасность.
2.4. Специфическая фармакологическая активность.
2.5. Фармакокинетические исследования [1].
В соответствии с правилами проведения доклинических исследований FDA, не существует «универсального» объема доклинических исследований. План доклинического исследования должен быть приспособлен к конкретным исследуемым веществам. Хотя правила и положения FDA не назначают стандартный набор тестов для всех исследуемых веществ, FDA выпустило рекомендации по выбору набора доклинических исследований [2,3].
1. Миронов А.Н. Бунатян Н.Д. и др. Руководство по проведению доклинических исследований лекарственных средств. Часть первая
2. U.S. Food and Drug Administration Center for Drug Evaluation and Research Handbook
3. S6 Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals (1997)
Последние видео 24.03.2016 VII научно-практическая конференция «Актуальные проблемы оценки безопасности лекарственных средств»24.03.2016 VII научно-практическая конференция «Актуальные проблемы оценки безопасности лекарственных средств», организованная НИИ фармации ГБОУ ВПО Первый МГМУ имени М.И. Сеченова МЗ России и секцией лекарственной токсикологии Российского общества токсикологов.
Staircase testУстановка «Staircase test» предназначена для изучения моторики передних конечностей у крыс.
Тест цилиндрТест «Цилиндр» используется для выявления асимметрии использования передних конечностей, например, при одностороннем повреждении сенсомоторной коры головного мозга крысы.
Напишите нам
Доклинические исследования занимают важное место в ходе разработки и изучения лекарственных средств. В регистрационном досье в формате Общего технического документа (Common Technical Document — CTD) отчеты об этих исследованиях приводят в модуле 4. В Европейском Союзе имеется стройная система законодательных и нормативных положений относительно доклинических исследований, которые изложены, главным образом, в следующих документах: Директиве 2001/83/ЕС с поправками, Директиве Комиссии 2003/63/ЕС, Директиве 2004/10/ЕС, документе «Notice to Applicants» (v. 2A «Procedures for marketing authorization. Chapter 1. Marketing authorization»), а также в многочисленных руководствах по доклиническим и клиническим исследованиям.
При ознакомлении с Порядком становится очевидным, что этот документ не является гармонизированным с каким-либо из указанных европейских документов, поскольку имеются изменения в техническом содержании и структуре, изменения четко не определены, отсутствует четкое соответствие между Порядком и каким-либо из европейских документов. Таким образом, Порядок неэквивалентен документам ЕС и его введение не означает принятия европейских норм. Совпадение отдельных положений не изменяет ситуации, поскольку в ЕС эти положения действительны в рамках всесторонней системы нормативно-правовых актов и нормативных документов, которая отсутствует в Украине.
Порядок регламентирует объем доклинических исследований (раздел IV) для следующих лекарственных средств:
1. Нового действующего вещества;
2. Нового вспомогательного вещества;
3. Фиксированных комбинаций;
4. Растительных лекарственных средств.
В п. 1 дважды также упоминается готовая лекарственная форма. При этом не оговорено, какие исследования необходимо проводить для действующего вещества, а какие для лекарственного препарата.
В Порядке не оговорены необходимость и особенности проведения доклинических испытаний других видов лекарственных средств, указанных в приложении 1 к Директиве Комиссии 2003/63/ЕС. К этим препаратам относятся: препараты с хорошо изученным медицинским применением, по существу аналогичные лекарственные средства (препараты-генерики), аналогичные биологические препараты, препараты из плазмы крови, вакцины, радиофармацевтические и гомеопатические препараты, препараты-сироты, лекарственные средства для передовой терапии и т.д. Также не рассматриваются ограниченные доклинические исследования в случае предоставления смешанных заявок (mixed marketing authorization application), которые в ЕС регламентируются Руководством CPMP/SVP/79995 «Guideline on non-clinical documentation for mixed marketing authorization applications».
Много вопросов вызывает отсутствие в Порядке информации о доклинических исследованиях препаратов-генериков, которые занимают ведущее место в номенклатуре украинских заводов, что можно истолковать как отсутствие необходимости в их проведении в любой ситуации. Для однозначного понимания этого вопроса рассмотрим положения о препаратах-генериках, принятые в ЕС.
В ст. 10(2)(b) Директивы 2001/83/ЕС с поправками, внесенными Директивой 2004/27/ЕС, дано следующее определение препарата-генерика:
«Генерический лекарственный препарат — это лекарственный препарат, который имеет тот же качественный и количественный состав действующих веществ и такую же лекарственную форму, как и референтный лекарственный препарат, и в целом биоэквивалентен референтному препарату. что доказано соответствующими исследованиями биодоступности. Различные соли, эфиры, сложные эфиры, изомеры, смеси изомеров, комплексные соединения или производные действующего вещества могут рассматриваться как такое же действующее вещество, за исключением тех, которые имеют значительные отличия в свойствах в отношении безопасности и/или эффективности. В таких случаях заявитель должен представить дополнительную информацию, доказывающую безопасность и/или эффективность разных солей, эфиров или производных утвержденного лекарственного вещества. Различные лекарственные формы для орального применения с немедленным высвобождением могут рассматриваться как одна и та же лекарственная форма. Исследования биодоступности могут не потребоваться от заявителя, если может быть доказано, что генерический лекарственный препарат отвечает соответствующим критериям, указанным в определенных детальных руководствах».
В этом определении заложены три фундаментальных признака генерических препаратов, на которые обращено внимание также в документе «Notice to Applicants» (т. 2A, глава 1, п. 5.3.2 «Generic medicinal product») и который отсылает заявителя к Руководству по исследованию биодоступности и биоэквивалентности.
В ст. 10(3) Директивы 2001/83/ЕС с поправками, внесенными Директивой 2004/27/ЕС, указано:
«Если лекарственный препарат не соответствует определению генерического лекарственного препарата, как указано в п. 2(b), или если биоэквивалентность не может быть доказана с помощью исследований биодоступности. или если имеются изменения действующего вещества, терапевтических показаний, силы действия, лекарственной формы или пути введения по сравнению с референтным лекарственным препаратом, должны быть представлены результаты соответствующих доклиническихили клинических исследований».
Приведенные положения однозначно свидетельствуют о следующем:
во-первых, для генерических лекарственных препаратов в ЕС в определенных случаях на законодательном уровне предусмотрено проведение доклинических исследований;
во-вторых, препараты-генерики могут быть не полностью идентичны референтному препарату; они могут иметь по отношению к нему определенные изменения, в том числе и в составе вспомогательных веществ;
в-третьих, из ст. 10(3) очевидно, что доклинические исследования в ряде случаев могут быть альтернативой клиническим испытаниям.
Обратимся по поводу доклинических исследований к Руководству CPMP/EWP/QWP/1401/98 «Note for Guidance on the Investigation of Bioavailability and Bioequivalence» (2001), к которому отсылает заявителей документ «Notice to Applicants». В определении «биоэквивалентность » указано: «…В качестве альтернативы классическим исследованиям биодоступности с использованием фармакокинетических конечных точек для оценки биоэквивалентности можно предусмотреть другие виды исследований, например исследования на людях с использованием клинических или фармакодинамических конечных точек, исследования на животных с использованием моделей или исследования in vitro. если они являются надлежащим образом обоснованными и/или валидированными».
Далее в Руководстве указано:
«5.1.8 Препараты для местного применения
а) местного действия
Для препаратов для местного применения (при оральном, назальном, ингаляционном, глазном, накожном, ректальном, вагинальном и других путях введения), действующих без всасывания в системных кровоток, не применим способ определения биоэквивалентности, основанный на определении в системном кровотоке. В принципе, необходимы фармакодинамические или сравнительные клинические исследования. Отсутствие таких исследований должно быть обосновано (см. соответствующие руководящие указания, в частности «Note for Guidance on clinical requirements for locally applied, locally acting products containing known constituents»).
Если системное воздействие, вызванное лекарственными препаратами для местного применения и местного действия, влечет за собой риск системных побочных реакций, системное воздействие необходимо определять».
Для определения системного воздействия, конечно, необходимо проведение доклинических токсикологических исследований.
20 января 2010 г. Европейское агентство по лекарственным средствам (European Medicines Agency) приняло новую версию Руководства CPMP/EWP/QWP/1401/98, которая вводится в действие в ЕС с 01 августа 2010 г. И в версии 2001 г. и в версии 2010 г. относительно лекарственных препаратов местного действия приведена ссылка на нормативный документ CPMP/EWP/239/95 final «Note for guidance on clinical requirements for locally applied, locally acting products containing known constituents» («Руководящие указания в отношении требований к клиническим испытаниям препаратов для местного применения, оказывающих местное действие и содержащих известные компоненты»), перевод которого на русский язык представлен ниже. В этом документе указано, что на препараты местного действия концепция аналогичности по существу, требующая определения биодоступности и не предусматривающая фармакологических и токсикологических исследований, не распространяется. В нем предусмотрены исследования на экспериментальных животных препаратов местного действия, в частности препаратов-генериков, а также препаратов с изменением в спецификации действующего вещества относительно его физических свойств, изменением в составе вспомогательных веществ или изменением устройства для применения. При этом фармакодинамические исследования или исследования местной доступности, или исследования in vitro могут рассматриваться в качестве альтернативы клиническим испытаниям.
Таким образом, Порядок, не предусматривающий проведения доклинических исследований при создании препаратов-генериков местного действия, а также в случае внесения определенных изменений в лекарственные препараты для местного применения, не учитывает соответствующие законодательные и нормативные положения ЕС. В этом отношении он вступает в противоречие также и с нормативными документами, принятыми в Украине, в частности с Руководством 42–7.1:2005 «Лекарственные средства. Исследование биодоступности и биоэквивалентности», которое гармонизировано с Руководством CPMP/EWP/QWP/1401/98 «Note for Guidance on the Investigation of Bioavailability and Bioequivalence» (2001) и введено в действие приказом МЗ Украины от 25.04.2005 г. № 191. Кроме того, вновь принятый Порядок не соответствует положениям нормативного документа «Порядок направлення на додаткові випробування лікарських засобів при проведенні експертизи реєстраційних матеріалів», утвержденного приказом МЗ Украины от 17.04.2007 г. № 190. Последний в п. 3.2(д) приложения предусматривает, что эквивалентность препаратов не в виде растворов, несистемного действия (например для орального, назального, офтальмологического, дерматологического, ректального или вагинального применения) и без системной абсорбции доказывают путем проведения, например, сравнительных клинических или фармакодинамических, дерматофармакокинетических исследований и/или исследований in vitro.
В связи с тем что в ЕС доклинические исследования требуются также при внесении изменений, рассмотрим действующие в ЕС и Украине положения о внесении изменений в сравнительном аспекте.
В ЕС в отношении оценки изменений в условиях торговой лицензии на лекарственные препараты для человека и лекарственные препараты для ветеринарии приняты два постановления: Comission Regulation (EC) № 1084/2003 и Comission Regulation (EC) № 1085/2003; кроме того, принят документ «Guideline on Dossier Requirements for Type IA and IB Notifications» («Руководство по требованиям к досье для уведомлений типа IA и IБ»). МЗ Украины принят приказ от 01.03.2006 г. № 95; приложение 5 к этому приказу регламентирует требования к документам при внесении изменений типа IA и IБ в регистрационные материалы и в целом гармонизировано с соответствующим руководством ЕС. В приложении 6 приведен перечень изменений, которые требуют новой регистрации лекарственного средства. В таблице приведено сравнение этих изменений с изменениями, требующими в ЕС не новой регистрации, а расширения заявки .
В ЕС при внесении указанных изменений сохраняется торговая лицензия (регистрация) и название лекарственного средства. В Украине при аналогичных изменениях и добавлении показаний в другой терапевтической области требуется новая регистрация, а не расширение регистрационного досье с представлением соответствующих материалов.
Существенные изменения в ЕС и в Украине
3. Зміни дозування, лікарської форми та способу застосування:
Не все приведенные изменения в таблице гармонизированы с европейскими нормами. К категории изменений отнесли добавление одного или нескольких действующих веществ, исключение одного или нескольких действующих веществ, количественные изменения действующих веществ ; но ведь это не изменения, а создание нового оригинального препарата или фиксированной комбинации, что требует в соответствии со ст. 8(3)(i) и 10b Директивы 2001/83/ЕС предоставлять результаты фармацевтических (физико-химических, биологических или микробиологических), доклинических (токсикологических и фармакологических) и/или клинических испытаний. Однако ни приложение 6 к приказу от 01.03.2006 г. № 95, ни Порядок, введенный приказом от 14.12.2009 г. № 944, не регламентируют, какие при этих изменениях необходимо проводить исследования и какие соблюдать условия, как это сделано для несущественных изменений типа IA и IБ. В ряде случаев при таких изменениях, которые ранее назывались изменениями типа II, может потребоваться проведение доклинических исследований (см. ст. 10(3) Директивы 2001/83/ЕС с поправками, внесенными Директивой 2004/27/ЕС, и Руководство CPMP/EWP/239/95 final).
Необходимо понимать, что в ЕС действует и постоянно актуализируется целостная система нормативно-правовой и нормативной документации, которой нет в Украине. Выхватывание отдельных положений при отсутствии других элементов системы может привести к негативным последствиям. Можно привести следующий пример. В Украине введено регистрационное досье в формате CTD. Для препаратов-генериков системного действия требуется полноценный модуль 3 «Качество» и подтверждение биоэквивалентности на основании сравнительного исследования биодоступности; фармакологические и токсикологические исследования не требуются. Однако в Украине почти отсутствует методологическая основа для проведения адекватных исследований по составлению модуля 3; введено всего 6 руководств по качеству, 4 из которых в настоящее время требуют актуализации. В то же время в ЕС введено и находится в стадии разработки около 80 руководств по качеству. В Украине не введено ни одного гармонизированного руководства по биологическим/биотехнологическим препаратам, в то время как в ЕС введено более 70 руководств и около 140 статей и монографий Европейской Фармакопеи по биологическим/биотехнологическим препаратам. В Украине сделана попытка регламентировать доклинические исследования путем введения Порядка, а в ЕС наряду с директивами введено в действие и находится на этапе разработки более 60 руководств по доклиническим исследованиям, которые регламентируются также и в некоторых из 180 руководств по клиническим испытаниям.
В принятом Порядке приведены положения, сочетающие нормативно-правовые требования, информационные материалы, инструкции и методические указания по выполнению тех или иных исследований и оформлению документов. Если в директивах и руководствах ЕС положения относительно доклинических исследований гармонично связаны с положениями относительно фармацевтических и клинических исследований, то в Порядке доклинические исследования рассматриваются вне общего контекста. При отсутствии Закона Украины «О лекарственных средствах», гармонизированного с директивами ЕС (в частности с Директивой 2001/83/ЕС с поправками), и необходимого комплекта руководств по доклиническим и клиническим исследованиям некоторые положения могут привести к неправильному планированию разработки в целом и неоднозначной оценке на этапе экспертизы.
На наш взгляд, в Порядке следовало представить цельный комплекс нормативно-правовых положений Директивы 2001/83/ЕС с поправками и других директив, которые в том числе касаются и доклинических испытаний.
Желательно к Порядку предусмотреть «Предисловие», в котором указать, каким международным документам соответствуют его положения; наличие ссылок на директивы и руководства ЕС/ICH позволило бы соблюсти международные правила стандартизации и принцип прозрачности при принятии национального документа. Отсутствие ссылок на первоисточники и достаточно отрывочная информация рождает вопросы относительно корректности терминов, а также целого ряда положений. Например, в п. 11 (глава II) указано: «У випадку розкладу лікарського засобу в процесі його зберігання обов’язково проводяться токсикологічні дослідження продуктів розкладу, що можуть утворюватися». При этом не указаны нормативные документы, в соответствии с которыми следует проводить квалификацию примесей.
Не рассматривая все несоответствия в Порядке, можно отметить следующее:
Указанный Порядок и приложение 6 к приказу от 01.03.2006 г. № 95 во многом не соответствуют международным нормам и стратегии правительства Украины относительно интеграции в ЕС.
Учитывая вышеизложенное, считаем целесообразным отозвать приказ МЗ Украины от 14.12.2009 г. № 944, а также пересмотреть приложение 6 к приказу МЗ Украины от 01.03.2006 г. № 95 и гармонизировать указанные документы с соответствующими нормами ЕС.
Прецеденты принятия таких документов, как Порядок и приложение 6, во многом обусловлены нарушением правил системы стандартизации МЗ Украины (см. СТ МОЗУ 42–1.0:2005). Эти документы были приняты без широкого обсуждения с заинтересованными лицами, без рецензирования и составления сводки замечаний.
В этой связи было бы полезным возобновить работу Совета по стандартизации МЗ Украины и запланировать разработку и введение в действие целостной системы гармонизированных нормативных документов, в том числе регламентирующих и доклинические исследования.
Еuropean medicines Agency
Европейское агентство по лекарственным средствам
Отдел по оценке лекарств для человека
Руководящие указания в отношении требований к клиническим испытаниям препаратов для местного применения, оказывающих местное действие и содержащих известные компоненты
Требования к клиническим испытаниям препаратов для местного применения, оказывающих местное действие и содержащих известные компоненты
[статус нормативного документа Европейского агентства по лекарственным средствам с ноября 1995 г.]
Данное руководство следует рассматривать совместно с Директивой 65/65/ЕЕС с поправками, Руководящими указаниями в отношении сокращенных заявок (III/3879/90), Указаниями для заявителей в отношении новых систем и, если это имеет отношение к делу, Руководящими указаниями по доклиническому исследованию местной переносимости лекарственных препаратов.
В данных Руководящих указаниях определены требования к клиническим испытаниям (части IC3, IV досье) в отношении препаратов для местного применения, оказывающих местное действие и содержащих известное действующее вещество.
Препаратами, оказывающими местное действие, являются препараты, которые наносят местно и которые проявляют свое действие в месте аппликации; системное действие, если таковое имеется, следует рассматривать как нежелательный эффект. Примерами являются препараты для применения в дерматологии (например кремы, мази), ингаляционные препараты, такие как порошки или аэрозоли для ингаляции, глазные капли, ушные капли, назальные препараты, а также препараты для орального, вагинального или ректального применения, которые оказывают местное действие (см. также документ «Note for Guidance CPMP list for allowed terms III/3593/91»).
Для таких препаратов изменение в составе или лекарственной форме может повлиять на эффективность и/или безопасность. Это может происходить, например, при изменении физико-химических свойств препарата или при изменении неактивных ингредиентов и в связи с этим изменении степени пенетрации активного компонента. Более того, по меньшей мере в дерматологии основа-носитель сама по себе может оказывать влияние на заболевание.
Никакие из подобных препаратов нельзя рассматривать как аналогичные по существу. Однако, как правило, не требуется данных полных токсикологических и клинических исследований. Сокращенные заявки для препаратов местного действия следует, таким образом, относить к смешанным заявкам. Отсутствие данных должно быть обосновано в отчетах экспертов. Также в случае изменений II типа должна быть обеспечена терапевтическая эквивалентность с оригинальным препаратом. Выбор исследований и отсутствие данных должны быть обоснованы в отчетах экспертов.
Заявка в отношении препарата для местного применения с известным действующим веществом, ранее не применявшимся местно
В отношении клинической части досье в зависимости от препарата может требоваться либо полное досье, либо соответствующие комплексные исследования.
Как и в случае препаратов, оказывающих системное действие, в отношении препаратов для местного действия необходимо показать, что заявляемый препарат (либо генерик, либо препарат с изменениями в составе) является терапевтически эквивалентным уже зарегистрированному препарату, то есть, что оба препарата «равнозначны» в отношении эффективности и безопасности до такой степени, что данные, полученные для «инновационного» препарата (то есть, препарата, для которого получена торговая лицензия на основании досье с полным комплектом документации) применимы также ко второму препарату.
Для доказательства терапевтической эквивалентности, в принципе, необходимы клинические испытания, особенно в отношении препаратов для местного применения в дерматологии, однако могут быть использованы или разработаны другие модели. Для этого в зависимости от ситуации могут быть рассмотрены фармакодинамические исследования на человеке, исследования местной доступности или, если приемлемо, такие же исследования на животных или in vitro при условии, что все исследования адекватно валидированы. Более того, надлежащее внимание следует уделить безопасности и местной переносимости.
Важно отметить, что необходимо заранее определить критерии и нормы «эквивалентности» и что перед испытанием с использованием соответствующих статистических методов в протоколе следует установить количество пациентов. Пределы, в диапазоне которых препарат все еще считается «эквивалентным», зависят от исследуемого соединения и используемых параметров; таким образом, ссылка на руководство по биоэквивалентности, где отклонение 20% в большинстве случаев является допустимым, по существу неприемлема.
Также, чтобы показать терапевтическую эквивалентность, недостаточно как такового отличия от плацебо. В зависимости от ожидаемого ответа на плацебо и необходимости «отрицательного» контроля могут понадобиться сравнительные исследования в трех группах с использованием плацебо, нового препарата и уже зарегистрированного препарата; тем не менее, может быть достаточно и сравнительных испытаний в двух группах (см. руководство по биостатистике). Выбор плана исследований должен быть обоснован.
Если для доказательства терапевтической эквивалентности взамен клинических исследований выбрана другая модель, то следует показать/обосновать релевантность этой модели, то есть модель должна быть валидирована: в частности должна быть доказана ее взаимосвязь с терапевтической ситуацией. Следует отметить, что на данный момент имеется очень немного моделей, и большинство из них пока недостаточно валидированы.
В отношении препаратов для местного применения исследования биоэквивалентности, как правило, не являются подходящим способом для доказательства терапевтической эквивалентности, поскольку концентрации в плазме крови не имеют значения для местной эффективности, хотя они могут играть роль в отношении безопасности.
Как правило, безопасность и местная переносимость должны быть гарантированы знаниями о действующем веществе и выбором известных неактивных ингредиентов; тем не менее могут быть необходимы определенные дополнительные исследования препарата в целом (как смеси действующего вещества и всех неактивных ингредиентов) как на животных, так и на человеке.
Если ожидается или доказано, что количество действующего вещества в месте действия и/или в системном кровотоке достигает более высокого уровня, чем в случае оригинального препарата, необходимо представить соответствующие токсикологические данные и данные изучения безопасности на человеке. В некоторых случаях может быть принята аргументация относительно отсутствия таких данных, что должно быть обосновано в отчете эксперта.
Кроме того, если неактивный ингредиент впервые используется в составе лекарственных средств, требуются соответствующие данные токсикологических и фармакологических исследований, а также данные исследования безопасности на человеке, чтобы показать, что соединение действительно неактивно и безопасно, и определить его взаимодействие с активным ингредиентом, если таковое имеет место.
Все эти ситуации должны быть рассмотрены в отчетах экспертов; также должен быть обоснован выбор данных или их отсутствие.
Более того, при оценке риска со стороны нового препарата следует принимать во внимание тот факт, что само заболевание может влиять на абсорбцию/пенетрацию местно применяемого вещества и, следовательно, необходимы данные по безопасности при применении у пациентов.
Если определенные заявления сделаны в отношении препарата с измененным составом, они должны быть подтверждены соответствующими данными.
Если изменение в составе не требует новой (сокращенной) заявки, но его можно отнести к изменению II типа, необходимо рассмотреть терапевтическую эквивалентность, включая при необходимости местную переносимость. Как правило, требуются такие же данные, которые указаны выше для сокращенной заявки. В случае незначительных изменений могут быть приемлемы доводы в отношении того, что в таких данных нет необходимости. Однако это должно быть обосновано в отчетах экспертов.
Требования к клинической части досье в отношении препаратов для местного применения/местного действия
I. Известное действующее вещество, ранее не применявшееся местно
Полное досье или соответствующие комплексные исследования
Рекомендуемые новостиРедакция журнала «Фармакокинетика и Фармакодинамика» не несёт ответственность за содержание и достоверность рекламных материалов. Перепечатка опубликованных материалов разрешается только по письменному разрешению ООО «Издательство ОКИ» и согласованием с Редакцией журнала. Мнение Редакции может не всегда совпадать с мнением авторов. При копировании на сайт статей из журнала «Фармакокинетика и Фармакодинамика» активная ссылка на: http://PharmacoKinetica.ru/ обязательна! Публикации на PharmacoKinetica.ru не должны использоваться для самостоятельной диагностики и лечения, не должны рассматриваться в качестве рекомендаций пациентам и не могут служить заменой консультации врача.
© 2015 г. Все права на сайт и его содержимое принадлежат ООО «Издательство ОКИ» | Юридическая поддержка и бухгалтерское обслуживание осуществляется компанией «Фиолент»







